 Soon to be sophmore Rei Imada (CMC '20) has sequenced 3 gene regions for 89 xeniids this summer. Soon to be sophmore Rei Imada (CMC '20) has sequenced 3 gene regions for 89 xeniids this summer. For the past couple of weeks in the McFadden lab, I have been working on DNA extractions and gene amplifications for 89 samples of Xeniidae sent to us from the Queensland Museum in Australia. The samples were collected at three sites on the Great Barrier Reef, as well as from Western Australia. Xeniidae corals are a family of soft corals that reproduce at an extremely high rate, and are known to overpopulate areas where coral bleaching has affected other corals, leaving little room for any repopulation. However, corals of the Xeniidae family cannot be reliably identified to species using morphology, thus, in the McFadden lab I have been working on amplifying DNA barcodes that can be utilized to help distinguish between species with more certainty. Using Polymerase Chain Reaction (PCR), I have been amplifying three specific genes. The genes include two mitochondrial genes---COI and mtMutS, and a nuclear ribosomal gene--- 28S. The amplified genes have been sequenced, and I am currently cleaning and combining all three genes to assemble an extended barcode for each sample. I will then be able to compare the barcodes from different samples and group the specimens according to threshold levels in their genetic makeup at the three genes. The program I will be using, MOTHUR, groups samples together into molecular operational taxonomic units (MOTUs) if they have less than a 0.3% average genetic distance. From this, we will be able to get an idea of the species diversity of Xeniidae off Australia. In the following weeks, I will be working on editing the gene sequences as well as troubleshooting samples in which gene amplification was unsuccessful.
Rei Imada, Claremont McKenna College, ‘20
0 Comments
 Alicia Pentico (HMC '19) working at the computer in the lab-finding and extracting mitochondrial genomes and rDNA from the UCE assembly data. Alicia Pentico (HMC '19) working at the computer in the lab-finding and extracting mitochondrial genomes and rDNA from the UCE assembly data. In the McFadden Lab, we have been studying coral phylogenetics, hoping to examine how past events have affected the evolutionary relationships between corals and their relatives, so we can better understand the effects of climate change. Over the past 5 weeks, we have started building phylogenetic trees from a) mitochondrial DNA b) Ultraconserved Elements (UCEs) and c) nuclear rDNA for different coral samples. The nuclear rDNA and mitochondrial DNA from which those trees are being developed are byproducts of the UCE assembly data. With some UCE test run data from 16 octocorals, I have extracted primarily complete mitochondrial genomes. These genomes were annotated using MITOS. When the annotated results were not complete, and genes were missing or fragmented, I returned to the original genomes to find and extract the genes. These genes were then aligned using MAFFT and concatenated. Phylogenies are going to be created from these alignments using RAxML. For the UCEs, I have prepped another 72 samples for sequencing using Kapa HyperPrep kits.
We will keep you updated! Alicia Pentico (Harvey Mudd ’19) Cathy and Andrea recently participated in the MASS program through Scripps Academy (http://www.scrippscollege.edu/academy/math-and-science-scholars). This program is designed to connect high school girls with faculty at the Claremont Colleges. Faculty work closely with students on a research project, with a final culmination in a research presentation. Students travelled from high schools in Los Angeles to Claremont on four Saturdays during the fall. They were trained in DNA extraction, PCR amplification, Gel Electrophoresis, DNA alignment and Phylogenetic Analysis. The students also learned a bit about deep-sea corals! It was a fun experience, and we look forward to participating in the program again next year~
Two of our undergraduate summer researchers presented their research at the annual Summer Research Poster Celebration at Harvey Mudd College this month. Thanks again to Aaron Friend (senior), who spent the summer at the AMNH, and MIke Adams (junior) who spent the summer at HMC working on species delimitation of corals.  Senior HMC student Aaron Friend presents his work on black corals, which he conducted at the AMNH. 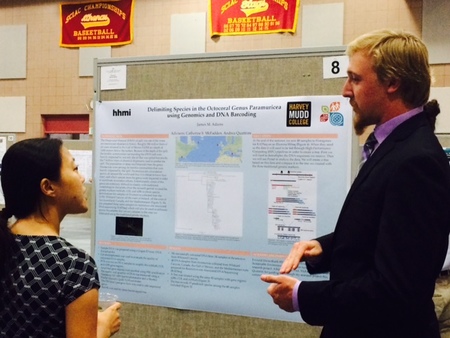 Junior Mike Adams discusses his research on Paramuricea species boundaries with faculty at the poster celebration. 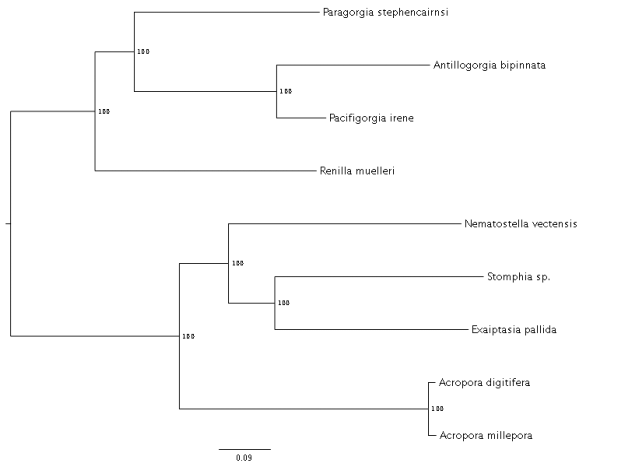 In Silico Target Capture of UCE loci based on an incomplete matrix (50%) of 721 UCEs. Genomic data only included in this analysis. In Silico Target Capture of UCE loci based on an incomplete matrix (50%) of 721 UCEs. Genomic data only included in this analysis. We have successfully designed 16,449 probes to target 1,795 loci across the anthozoa. We used several existing genomic and transcriptomic resources as well as our newly assembled Renilla muelleri genome (hybrid assembly). Brant Faircloth's amazing package Phyluce was used primarily to design these probes. Probes are being synthesized by MycroArray and we plan to test them on a subset of taxa before finalizing the probe set.. We will make the probe sequences publicly available.
And, we thank all of our collaborators who have provided essential data for use in probe design.  Craig in his PPE on the deck of the ship in the Gulf of Mexico. Thanks for your hard work this summer! Craig in his PPE on the deck of the ship in the Gulf of Mexico. Thanks for your hard work this summer! My name is Craig Dawes and I am an NSF-REU Scholar at the American Museum of Natural History, in New York City, working under the supervision of Dr. Estefania Rodriguez (Associate Curator of Marine Invertebrates) and Mercer R. Brugler (Assistant Professor at NYC College of Technology [CUNY]). I am also a full-time student in the Biomedical Informatics program at NYC College of Technology in Brooklyn, NY. I have been working in Dr. Brugler’s deep-sea molecular lab since January 2015, initially as a part of the Emerging Scholars program and then as an LSAMP (Louis Stokes Alliance for Minority Participation) Scholar. Based on my experience in the lab, Dr. Brugler recently placed me in a mentoring role; i.e., I am teaching new students how to extract and quantify DNA, set up PCR, visualize PCR on an agarose gel, set up a cycle sequencing reaction, and obtain DNA sequence data using a traditional ABI-3730xL Sanger sequencer. I am originally from Jamaica and moved to NYC about six years ago to pursue a degree in Nursing. After taking a Biology course with Dr. Brugler I was inspired to explore research as a career option.
My NSF-REU summer internship includes three projects: 1. I participated in a NOAA-funded ocean-going research expedition during Summer 2015 to the Flower Garden Banks National Marine Sanctuary in the Gulf of Mexico to collect mesophotic black corals. Mesophotic corals are defined as those organisms living in the middle of the photic zone, i.e. areas of low light penetration. We collected a total of 25 black corals representing three families and six genera across a depth range of 64 - 157 meters. Using three mitochondrial intergenic regions and three nuclear genes, I am obtaining a molecular barcode for these corals, in an effort to elucidate any undescribed species and/or extend the range of known species. We also surveyed banks within the sanctuary for Acanthopathes thyoides and Elatopathes abietina. 2. Based on morphology, Acanthopathes and Elatopathes are currently classified in the same family; however, they do not group together in a molecular phylogeny. These species are considered ‘wandering taxa’ as they change position depending on the gene (mitochondrial v. nuclear) or algorithm (Parsimony v. Likelihood v. Bayesian) used to build the phylogeny. We successfully collected two A. cf. thyoides and six E. cf. abietina. Elucidating 1) intraspecific variability within A. thyoides and E. abietina or 2) closely related cryptic species could potentially stabilize their phylogenetic position. 3. We recently obtained tissue samples from ten black corals that were collected during the 2015 Hohonu Moana Expedition (aboard the NOAA ship Okeanos Explorer) that explored deep waters surrounding the Hawaiian Archipelago. Dr. Dennis Opresko (Smithsonian NMNH), the world’s foremost expert on black coral taxonomy and systematics, noted that several individuals might be new to science based on a rough morphological examination. Thus, I am also barcoding these samples using mitochondrial and nuclear DNA in hopes of elucidating potentially new species. Other projects - Molecular characterization of Deep-Sea “Sea Anemones” from the Arctic Ocean We also obtained three specimens from Beaufort Sea, outlying the Arctic Ocean, at a depth of 1000m. Two of these specimens were tentatively identified as Kadosactis rosea, Allantactis parasitica and an unknown species presumed to be a member of the order Actiniaria. We amplified three mitochondrial, genetic markers to confirm the morphological identification of the first two specimens and reveal the identity of the unknown specimen. Our DNA analysis of the unknown suggests that we may have found a representative of a new genus. Currently I am analyzing the morphology of the animal via histological and microscopic examination. Future work will place these three specimens in a phylogenetic context.  Pomona undergrad Thomas Byrne (18') prepping octocoral DNA for PCR. Thanks for your hard work this summer, Thomas! Pomona undergrad Thomas Byrne (18') prepping octocoral DNA for PCR. Thanks for your hard work this summer, Thomas! For the past several weeks we have been busy preparing deep-sea octocorals for multi-locus DNA Barcoding from our recent RV Celtic Explorer voyage to Whittard Canyon. Initial identification of samples, based on morphology, indicates that the octocorals contributing to the diversity within Whittard Canyon are: Isididae, Plexauridae, Primnoidae, Acanthogorgiidae, Alcyoniidae, Chrysogorgiidae, Paragorgiidae, Clavulariidae, and Pennatulacea. So far, we have extracted high quality DNA from each sample and have amplified both the mtMutS and COI markers. We have recently begun amplifying a third marker, nuclear 28s rDNA. Additionally, we are amplifying the IGR4 region of the Isididae samples in order to further delineate taxonomic relationships. Once amplified, our samples will be sent off for sequencing; these sequences will then be edited and aligned in order to construct preliminary phylogenetic trees. Our ultimate goal with these markers is to better understand the species richness of Octocorallia in Whittard Canyon. Furthermore, we will be calculating genetic distances for between our samples and compare these to previously studied specimens from around the world in order to aid in our species identification and estimates of species richness. We hope to have our analyses completed in the coming few weeks!
Thomas Byrne, Pomona College '18  Harvey Mudd Junior Mike Adams taking a break from setting up a PCR to smile at the camera. Thanks for all your hard work this summer! Harvey Mudd Junior Mike Adams taking a break from setting up a PCR to smile at the camera. Thanks for all your hard work this summer! Over the past three weeks we have been working hard in the McFadden lab to prepare 47 Paramuricea samples for Restriction Site Associated DNA Sequencing (RADSeq). These samples come from Canada, the Gulf of Mexico, and Whittard Canyon off the coast of Ireland. Ultimately we wish to compare the phylogenetic tree we build from the RADSeq data to a tree built using genes that have been conventionally used for octocoral systematics to understand species boundaries in this genus. In order to do this we have successfully extracted high quality DNA from Paramuricea samples collected during the RV Celtic Explorer cruise that took place last month. We have also been working to amplify mtMutS and COI, and n28S from all Paramuricea samples collected off Ireland; those from the Gulf of Mexico and Canada are completed (see Doughty et al. 2014). Because the DNA from some of the older samples collected off Canada and in the Gulf are degraded, we will be re-extracting the DNA from original tissue samples over the next week or so. Plus, we just obtained tissue samples from Paramuricea in the Mediterranean. We hope to submit samples for RADSeq by the end of the month. We will update you on the results!
Mike Adams (Harvey Mudd '18) Excited to say that we have draft genome assemblies for nine anthozoans! I used bbmerge and bbduk (https://sourceforge.net/projects/bbmap/) to clean and trim sequences. DiscovarDenovo (https://www.broadinstitute.org/software/discovar/blog/) was used for the draft assemblies. Computation time on nine species was ~1 week (on a 512 GB RAM 64 processor). That is all!
We are currently comparing the draft assemblies to other assemblers (SOAPdenovo, SPAdES) and I am also blasting the contigs (and also the trimmed reads) for environmental contaminants. This latter step is the tricky part. If you are reading this and have any advice, please send it my way. Finally, we have sequenced Renilla on PacBio as well. I will be coupling these data with the Illumina data to generate a hyrbid assembly. Exciting! Stay tuned for more~ Andrea Happy Summer Solstice! Its been a great start to summer 2016. I was fortunate to spend 3 weeks off Ireland surveying deep-sea coral communities in Whittard Canyon. Check out http://scientistsatsea.blogspot.com/ for more information about the cruise (led by Louise Allcock at NUIG).. Beautiful weather and a lot of hard work by many (including several students) equalled a very successful cruise! Stay tuned for more blogs this summer! For now, a beautiful sunrise~  |
UCE Project TeamAll things Anthozoa, Evolution and Ecology Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation
Archives
September 2016
Categories |




 RSS Feed
RSS Feed
